Our Service
Overview
According to NMPA requirement, all the medical devices applying for certificate need to be classified based on the risk level, filing is required for Class I medical devices with low risk while Class II & III with higher risk need to go through the registration process under the administration of NMPA. Clinical evaluation is a mandatory process while whether clinical trial is necessitated depends on specific cases.
- Filing of Class I medical devices
- Initial Registration of Class II & III medical devices
- Renewal of Class II & III medical device Certificate
- Registration Change of Class II & III medical device Certificate
- Registration Document Preparation & Review
- Technical Requirement Drafting Support
- Product Testing
- Advisory Assistance and Guidance on Internal System Audit
- Supplementary Material Preparation Guidance
- Clinical Evaluation Strategy Support
Overview
To meet the compliance requirement of NMPA in the whole registration process and post registration process,
medical device manufacturers are necessitated to show commitment on compliance regarding inspections from
the government such as unannounced inspection, internal system audit and so forth.
- Simulated Pre-assessment for on-site Inspection in the Medical Device Initial Registration Stage
- Unannounced Inspection Simulation for Manufacturers after acquisition of Market Clearance of target medical device
- Advisory Assistance and Guidance on Internal System Audit
Overview
According to the requirements of Good Manufacturing Practice (GMP) and Good Supply Practice (GSP), enterprises are obliged to follow relevant requirements along with product characteristics to establish and improve the Quality Management System compatible with the medical devices produced and operated, ensuring effective operation. Regarding the risk of medical device, in order to manufacture quality-compliant products with safety and effectiveness, it is necessary to establish a systematic, coordinated, and effective quality system based on hardware, supported by the document system, and guaranteed by personnel.
- Consultancy Service on Clean Workshop Establishment and Verification
- Consultancy Service on the Process of Design and Development
- Consultancy Service on Preparation and Improvement of Quality System Documents.
- Consultancy Service on Common Equipment Verification
- Consultancy Service on Special Process Verification
- Compliance Review Support on QS Hardware
- Training support on Establishment and Execution of the Quality System.
- Support on Preparation, and Submission of QS Assessment Materials until the pass of the Assessment , and successful acquisition of the Assessment Report.
Overview
The MAH system stands for marketing authorization holder system that affects domestic MD registration. Other than MD manufacturers with manufacturing license, the provision of MAH provides compliance accessibility for independent medical institutions and scientific researcher of medical devices to apply for marketing authorization certificate without meeting the mandatory qualification of holding manufacturing license as previously required. The MAH is designed to untie the bundled regiment of the marketing clearance and manufacturing license under one entity in order to benefit patients through encouraging MD innovation by involving more R&D driven institutions and personnel without manufacturing capability, making both the MD researchers and manufacturers specialized in respective fields. Provided the MAH entrusts manufacturers the production of medical devices, the MAH has the statutory obligation to be legally responsible for the entire relevant procedure of MDs including R&D, clinical trials, manufacturing, marketing & distribution, and proper usage of MDs.
- For the quality system and risk system, providing the MAH with multi-dimensional assistance with professionalism during the registration process
- Promptly keeping clients informed regarding the latest Regulations of the Registrant System in different regions
- Assistance on matching suitable manufacturers for registrants.
- MAH planning involving bilateral bundling the systems of the R&D and the manufacturers
- Simulated GMP inspection further ensuring the success of the review
- Selected Suppliers ranging from entrusted manufacturers to product tests with credible service
Overview
According to the requirement of Regulations on the Supervision and Administration of Medical Devices (No.739 Decree of the State Council of China) , manufacturers specializing in Class II & III medical device production are obliged to apply for the Production License by meeting necessary compliance requirement of NMPA.
- Documents Preparation
- Assessment on Spot
- Application Material Review before Acceptance
- Advisory Service for Enterprise Rectification
- Notification after Approval
Overview
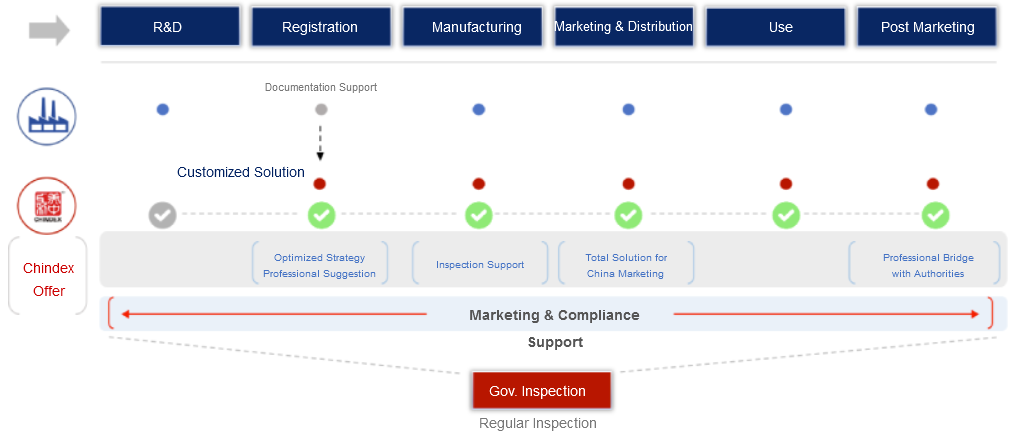
With the continuous development of Chinese medical device review system and the healthcare industry, more innovative medical devices with cutting-edged technology are introduced to China. International medical device manufacturers start to shift their strategic focus on the Chinese market. For the international MD manufacturers willing to conduct the initial move in China, appointing an agent in China is a must-do to meet statutory requirement. As a reliable partner with 30 years of medical device industry experience, Chindex has been committed to providing support engaging with Chinese authorities throughout the whole process and helping international medical device manufacturers to strengthen the business layout in the Chinese market .
Unfortunately, it is NOT legal unless the manufacturers have branches with qualification in China to deal with the registration issues. Otherwise, it is a must-do that manufacturers overseas are required to appoint a Chinese agent for the medical device registration.
Regarding the registration fee for NMPA, as for the imported medical devices, the registration of Class II medical device needs 210,900 CNY while that of Class III costs 308,800 CNY. In addition, other kinds of possible fees should be taken into consideration such as fees of document translation, clinical trial, and so forth.
After the registration materials are accepted by NMPA, technical review for Class II medical devices takes 60 working days to process while for Class III medical devices, technical review takes 90 working days. If supplementary materials are required by NMPA, manufacturers are obliged to conduct submission within one year, otherwise the review may result in termination.
MDs with CE or FDA certificate are feasible to enter SAR without NMPA certificate. As for the Greater Bay Area and Hainan province, MDs marketed in HK or MO as a premise can enter the pilot areas if designated medical institutions in pilot areas are willing to procure medical devices out of clinical urgent needs under the approval of local government.
Copyright © Chindex Medical Limited

